
Mechanism and dynamics of extracellular cellulolytic protein interactions
JOINT-SUPERVISORS: Roland LUDWIG, Chris OOSTENBRINK
Background.
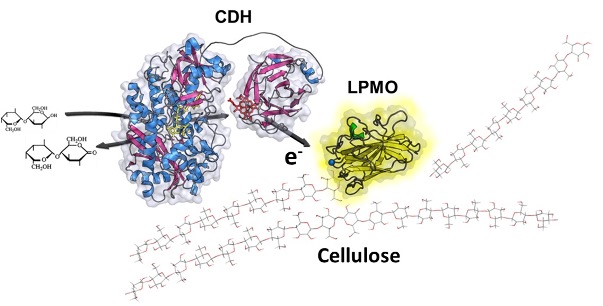
The need for renewable energy is growing rapidly and biofuel derived from lignocellulose is an attractive alternative to fossil-based fuels. Notwithstanding, the visionary green energy concept is challenged by the recalcitrant nature of the major component cellulose, which makes bioconversion to fermentable saccharides problematic and costly. Recently, an oxidative enzymatic system was discovered, where copper-dependent lytic polysaccharide monooxygenase (LPMO) cooperates with the extracellular flavocytochrome cellobiose dehydrogenase (CDH) to catalyse redox-mediated glycosidic bond cleavage in crystalline cellulose and hemicelluloses (Quinlan et al. 2011). The LPMO/CDH system was shown to enhance the degradation of cellulose by hydroxylation at C1 or C4 which leads to an elimination of the glycosidic bond (Beeson et al. 2012). The precise mechanism of the monooxygenation reaction is hereto unknown, but it involves reduction of the copper center of LPMO by CDH and the formation of a reactive oxygen species to initiate substrate hydroxylation, either by a superoxo (Li et al. 2012) or an oxyl (Kim et al. 2014) mechanism. To understand the catalytic mechanism of LPMO, the elucidation of its interaction with CDH and the dynamics of this interaction is indispensable. As a first step into this endeavour, the full structures of CDH and LPMO from N. crassa were elucidated in a collaboration of C. Divne (Karolinska Institute, Stockholm, Sweden) with LUDWIG (manuscript submitted). The CDH structures show open and closed CDH conformers, which emphasize the need to study domain interactions and the necessary electron transfer between domains.
The structure and function of complex macromolecular entities like proteins have been studied by molecular simulation and led to insight into the molecular mechanisms that underlie experimental observations (Karplus & Kuriyan, 2005; van Gunsteren et al., 2006). Sampling involves the generation of a sufficiently large amount of molecular structures to describe the orientations and conformations that may be obtained from a variety of search techniques, like systematic searches, molecular dynamics or Monte Carlo simulations. Scoring is required to evaluate the generated conformations. The assignment of an energy value to a molecular conformation can again be done by quantum mechanical calculations, through force fields to empirical, or knowledge-based scoring functions.
In order to elucidate the electron transfer process between CDH and LPMO, the atomic and possibly electronic degrees of freedom need to be taken into account, while an appropriate description of the protein interaction requires extensive sampling of the conformational space. In order to quantify the equilibrium between the interacting proteins, the interdomain interactions may be described in molecular simulations (OOSTENBRINK). Experimental proof will be obtained from fast-kinetic and calorimetric techniques in combination with rational protein engineering (LUDWIG).
Aims and methods.
In this project, we will study the protein interaction and the electron transfer between CDH and LPMO. A starting point for these studies is N. crassa CDH IIA and LPMO9F for which we deduced the structure with our partner C. Divne or other LPMOs with published structural data (Hemsworth et al., 2013). Based on the crystal structures of CDH and LPMO we propose a direct electron transfer between haem b in CDH and the copper in LPMO. However, the copper is centered in a flat surface of LPMO, which is facing the surface of cellulose during catalysis and thus possibly blocked. It will be of particular interest to determine whether alternative binding sites and electron pathways to the LPMO active site are feasible by using molecular modeling. We have recently successfully described the electron transfer paths in cytochrome P450 reductase, another multidomain electron transfer protein displaying an open and a closed state (Sündermann and Oostenbrink, 2013). To predict complexes of CDH with LPMO, we will use state of the art protein-protein docking experiments (Dominguez et al., 2003, De Vries et al., 2007). Subsequently, we plan to use the distance-field reaction coordinate, which we developed recently (De Ruiter and Oostenbrink, 2013), in conjunction with either Hamiltonian replica exchange simulations or with local elevation to optimize and quantify the strength of the interaction. The effect of protein dynamics on the efficiency of the electron transfer pathway will be studied. The electron transfer can be described classically using decay rates for different types of connections between atoms (Beratan et al., 1990) and the most likely paths can be obtained using Dijkstra's algorithm (Dijkstra, 1957). By performing this analysis on a large set of protein conformations the dynamic fluctuations in the electron transfer may be determined (OOSTENBRINK).
By constructing specific mutations that may influence the proposed interaction of CDH and LPMO or electron transfer pathway, the suggested function of the amino acids may be validated experimentally. Amino acids identified as important for CDH/LPMO interaction or electron transfer will be mutated to prove or disprove their importance. Enzyme variants will be produced in Pichia pastoris (Kittl et al. 2012, Sygmund et al. 2012) and studied by biochemical, bioelectrochemical and biophysical methods (LUDWIG) including rapid kinetics (determination of electron transfer rates) as well as EPR (domain mobility and movement, reduction of the type 2 copper center in LPMO, OBINGER) and isothermal calorimetry (enthalpic and entropic binding components).
Collaborations This thesis is envisioned as a joint project between OOSTENBRINK (molecular modelling and simulations) and LUDWIG (protein engineering and characterization). Collaborations within BioToP are foreseen with OBINGER (EPR). International collaborations include DIVNE (structural biology, crystallography)
Beeson, WT, Phillips, CM, Cate, JH, Marletta, MA (2012) J. Am. Chem. Soc. 134, 890-892
Beratan DN, Onuchic JN, Betts JN, Bowler BE, Gray HB (1990) J Am. Chem. Soc. 112, 7915-7921
de Ruiter A and Oostenbrink C (2013) J. Chem. Theory Comp. 9, 883-892
de Vries SJ, van Dijk ADJ, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AMJJ (2007) Proteins 69, 726-733
Dominguez C, Boelens R, Bonvin AMJJ (2003) J. Am. Chem. Soc. 125, 1731-1737
Dijkstra EW (1959) Numer Mathem 1, 269-271
Hemsworth GR, Davies GJ, Walton PH (2013) Curr. Opin. Struct. Biol. 23, 660-668
Karplus M, Kuriyan J. (2005) Proc. Nat. Acad. Sci. USA 102, 6679-6685
Kim S, Ståhlberg J, Sandgren M, Paton RS, Beckham GT (2014) Proc. Natl. Acad. Sci. USA 111, 149-154
Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R (2012) Biotechnol. Biofuels 5, 79
Li X, Beeson WT, Phillips CM, Marletta MA, Cate JHD Structure (2012) 20, 1051-1061
Quinlan RJ, Sweeney MD, Leggio LL, Otten H, Poulsen J-CN, Johansen KS, Krogh KBRM, Jørgensen CI, Tovborg M, Anthonsen A, Tryfonad T, Walter CP, Dupree P, Xua F, Davies GJ, Walton PH (2011) Proc. Natl. Acad. Sci. USA 108, 15079-15084
Suendermann A and Oostenbrink C (2013) Protein Sci. 22, 1183-1195
Sygmund C, Kracher D, Scheiblbrandner S, Zahma K, Felice AKG, Harreither W, Kittl R, Ludwig R (2012) Appl. Environ. Microbiol. 78, 6161-6171
van Gunsteren WF, Bakowies D, Baron R, Chandrasekhar I, Christen M, Daura X, Gee P, Geerke D, Glaettli A, Hnenberger PH, Kastenholz MA, Oostenbrink C, Schenk M, Trzesniak D, van der Vegt NFA, Yu HB (2006) Angew. Chem. Int. Ed. 45, 4064-4092