
Identification of both coding and non-coding genomic regions that contribute to Bioprocess relevant behaviour
SUPERVISOR: Nicole Borth
Background.
The CHO genome contains all relevant information to develop and maintain a hamster, consisting of a myriad of different cell and tissue types and stages of development, where all kind of diverse functions have to be fulfilled. In industrial production, however, cells are maintained under relatively constant and controlled culture conditions with reliable nutrient supply. All that is expected of them is to divide while producing and processing a recombinant protein. In this context it is also important to understand that the genome of mammalian species consists of less than 3% coding genes, while the rest has regulatory function: in part transcribed as non-coding RNAs such as miRNAs and lncRNAs, in part acting as cis- and trans-regulatory sequences, such as promoters, enhancers, silencers and similar. Very little is known about their precise mechanisms and most knockout studies so far were targeted against coding genes only. Finally, most annotations available for coding genes are derived from studies of embryonic development or from medicine – again withfocus on functionality in the context of the full organism rather than an in-vitro cultured cell line. Frequently functional annotations are found such as “nerve development”, however the associated gene is also expressed in other tissue types.
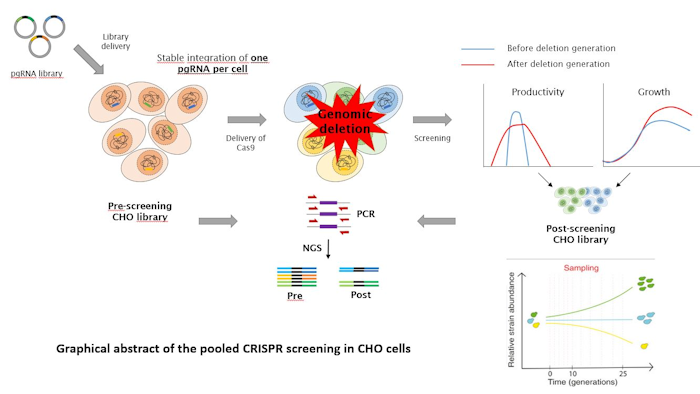
To knock-out such non-coding function, the traditional approach of generating a frameshift mutation that disrupts the sequence of the translated protein is not feasible. For this purpose, a deletion strategy was recently developed that enables removal of entire fragments of the genome up to 150 kb in size using paired gRNAs[1]. A pipeline for the design of such paired gRNAs is available and was tested on a small library targeting a subset of 500 lncRNAs[2]. Thus, the tools are available to start a genome wide deletion approach to understand the functionality of both coding and non-coding genomic regions in CHO cells under bioprocessing conditions.
Aims and methods.
The main objective of this project is the categorization of the genome into regions that are essential for growth under bioprocessing conditions and those that are dispensable. For this purpose, the 2,7 Gb genome of CHO will be classified in 100kb bins as i) essential (if deleted, cells will stop growing or die) or ii) non-essential (deletion has no impact on growth or survival). A novum here is that the entire genome will be investigated, including non-coding regions, which has not been attempted before.A detailed understanding of the mechanisms underlying the effect of deletion will be investigated on selected examples.
The initial approach is based on the design of a library of paired gRNAs that target 100kb segments of the entire genome. These gRNAs will be cloned into plasmids with the required promoter for transcription and integrated into the genome of CHO cells such that each cell will contain a single gRNA pair. After deletion, cells that continue to grow over a period of time will be analysed for those gRNA pairs that are still present in the population. Inherently, these will be for regions that are non-essential, while those gRNA pairs that are lost or reduced relative to the starting library can be assumed to be essential, as the deletion of the corresponding genomic region caused a stop in growth or cell death so that the corresponding gRNAs disappeared from the population.
Next, the genome segments will be screened for known coding and non-coding genes that might explain their essentiality. Particular focus will be given to genome regions that bear no known genes, where we will investigate the presence of unknown transcripts and chromatin state information, such as enhancers or silencers that may play a role. Also, smaller segments of a selected number of these genome regions will be deleted to narrow down the precise locus that is effective.
REFERENCES:
[1] Schmieder V, Bydlinski N, Strasser R, Baumann M, Kildegaard HF, Jadhav V, Borth N (2018) Enhanced genome editing tools for multigene-deletion knock-out approaches using paired CRISPR sgRNAs in CHO cells. Biotechnology Journal 13: 3: 1700211
[2] Schmieder V, Novak N, Dhiman H, Nguyen LN, Serafimova E, Klanert G, Baumann M, Faustrup Kildegaard H, Borth N (2019) An innovative pooled CRISPR/AsCpf1 screen tackling long non-coding RNAs in Chinese Hamster ovary cells. BMC Biotechnology submitted