
Structure and dynamics of cellobiose dehydrogenase and cytochrome P450
SUPERVISOR: CHRIS OOSTENBRINK
Project assigned to: GABOR NAGY
Background.
The structure and function of complex macromolecular entities, like proteins, polymers or mineral structures have been studied by molecular simulation and led to insight into the molecular mechanisms that underlie experimental observations (Karplus & Kuriyan, 2005; van Gunsteren et al., 2006). Two aspects are commonly distinguished that play crucial roles to get meaningful results: sampling and scoring. Sampling involves the generation of a sufficiently large amount of molecular structures to describe the orientations and conformations that in reality lead to the observed (average) quantities that are measured. These may be obtained from a variety of search techniques, like systematic searches, molecular dynamics or monte carlo simulations. Scoring, on the other hand, is required to evaluate how likely the generated conformations are in terms of energy and to discriminate ‘good’ conformations from ‘bad’ conformations. The assignment of an energy value to a molecular conformation can again be done in a variety of ways, ranging from advanced quantum mechanical calculations, through force fields to empirical, or knowledge based scoring functions.
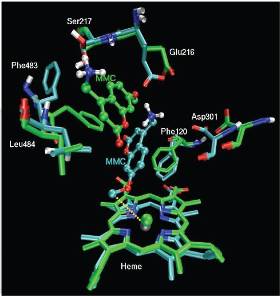
In this project the effect of structure and dynamics of experimentally investigated metalloproteins on various biophysical properties will be explored. Because of their special electronic properties, metalloproteins pose extra requirements on the scoring of conformations. We will describe the metal complexes either by including the electronic properties explicitly (using QM or hybrid QM/MM approaches) or implicitly by further optimizing available force fields that describe the interactions between atoms classically. In order to be able to rationalize, reproduce and predict experimental findings on these proteins, sufficient sampling of the protein conformational space is crucial.
Many of the studied properties of oxidoreductases studied in BioToP (see OBINGER and HALTRICH), like the redox potential, intramolecular electron transfer and catalysis of enzymatic reactions depend strongly on the structure and dynamics of the protein. Molecular modeling can play a vital role in the rationalization of a variety of biophysical properties that are experimentally accessible. In the past, we have attempted to calculate the redox potential of the copper-containing protein azurin (van den Bosch et al., 2005) and were able to explain findings in the Raman spectrum of the bacterial, heme-containing cytochrome P450 BM3 and several mutants (Stjernschantz et al., 2008). The effects of structural changes on metabolic activity of human cytochrome P450 enzymes were also studied extensively (De Graaf et al., 2007; Hritz et al., 2008).
Aims and methods.
In the context of this project, two proteins were selected. On the one hand we plan to study cellobiose dehydrogenase (CDH). This is a flavoheme protein, in which electrons are transferred from the substrate via the FAD and the heme to certain electron acceptors, but also to electrodes (Tasca et al., 2009; Coman et al., 2008). CDH is one of the few enzymes that can catalyse direct electron transfer to electrodes (without a mediator), and hence is of interest for biosensors, electrodes or biofuel cells. The aim is to improve the intramolecular electron transfer (from the flavo to the heme domain) and the transfer on to carbon electrodes. Our computational study of wild-type and mutant versions of this protein will be performed in close collaboration with HALTRICH, where this protein engineering study takes place experimentally.
A second class of protein targets is formed by heme-containing cytochrome P450 enzymes, of which the human form plays a role in the metabolism of xenobiotics (e.g. drugs) to facilitate excretion. Earlier studies have indicated that these proteins can perform their versatile functions thanks to a very malleable active site, accommodating very diverse substrates (Guengerich, 2006; Hritz et al., 2008). This active site flexibility places extra requirements on the sampling of the conformational space. Enhanced sampling techniques such as replica exchange molecular dynamics simulations may be used to describe the dynamics of the active site with sufficient detail (Hansmann, 1997; Sugita et al., 2000). Apart from a partner within the DK programme (OBINGER), a close collaboration is foreseen in this project with the group of Vermeulen (VU University, Amsterdam, The Netherlands), an internationally acknowledged expert in the field of cytochrome P450 research.
Degrees of freedom. An important question before undertaking any modeling project is the required level of detail. Especially in the case of metalloproteins, one should be aware of the special electronic properties of the complexes that play a role. In case where it is needed, we will use QM and QM/MM methods describing the electron density explicitly. In other cases, the electronic effects may be described more empirically, e.g. through force fields, which may need to be derived or refined. In collaboration with OBINGER, we would like to refine the currently available force field parameters for the heme group, to better reproduce experimental properties that depend on structural distortions of the heme group as observed in various complex structures.
Structure. The most ideal proteins to study computationally are those for which high-quality 3D structures are available, either from X-ray diffraction of protein crystals, or obtained from NMR spectroscopy. If structures for the proteins themselves are not available, reasonable models can be obtained on the basis of structures of homologous proteins. A recurring theme in this project will be the effect of specific mutations on the protein structure, stability and function. To study the effect of mutations, models can be constructed either statically, or based on a dynamic description of the protein in solution. We will create such models and explore the possibilities of mutated residues to be involved in modified interactions, to stabilize overall protein stability, or to have a local effect on the flexibility of neighbouring amino acids.
Dynamics. In many cases, it is not only the structure of a protein that is affected by a point-mutation, but also the dynamic behaviour of neighbouring amino acids changes. Since many processes within the protein are of a dynamic nature, this needs to be taken into account. The main method to be used will be molecular dynamics simulations, such that experimentally available quantities can be estimated as an ensemble average over the conformational space of the system. From the simulations, we can immediately monitor changes in fluctuations upon mutations, which can e.g. be observed through Raman (Stjernschantz et al., 2008), NMR (Keizers et al., 2005), or time-resolved fluorescence spectroscopy (Stortelder et al., 2006). Also when more empirical methods are employed, to e.g. describe electron transfer mechanisms at a low level, the effect of varying the protein conformation will be included.
In the first year of this project, the force field will be re-evaluated for use with the metalloproteins described here. Subsequently, we will study the structure and dynamics of the proteins and the relevant mutations in order to rationalize and reproduce available experimental data. The main aim of the project will be to be able to successfully predict the effect of specific point-mutations on the overall function of the selected target proteins.
Accurate calculations of enthalpy and entropy differences based on rigorous statistical mechanics for drug design are currently rather uncommon. This project will focus on such calculations, and will investigate the calculation of the ligand-surrounding enthalpies and entropies, as outlined above. The convergence characteristics of full entropy calculations versus ligand-surrounding entropies will be investigated. It is expected that the well-converging ligand-surrounding enthalpies and entropies will be better suitable to explain experimental findings in ligand binding. In the first year, different methods to calculate energies and entropies will be applied and compared. In the second year, special emphasis will be on the determination of the reduced properties. Special care will be taken to validate the methods against model systems and experimental data, in close collaboration with JUNGBAUER. Finally, the role of enhanced sampling techniques for these calculations will be explored in the second half of the project. The overall aim of this project is to develop methods that offer physically meaningful estimates of binding enthalpy and entropy without additional computational costs, as compared to free energy calculations. This highly valuable insight into these features of protein-ligand interactions can be practically attainable in relevant applications.
Coman, V., Vaz-Domínguez, C., Ludwig, R., Harreither, W., Haltrich, D., De Lacey, A. L., Ruzgas, T., Gorton, L., Shleev, S. (2008) Phys. Chem. Chem. Phys. 10, 6093-6096
De Graaf, C., Oostenbrink, C., Keizers, P. H. J., van Vugt-Lussenburg, B. M. A., van Waterschoot, R., Tschirret-Guth, R., Commandeur, J. N. M., Vermeulen, N. P. E. (2007) Curr. Drug Metab. 8, 59-77
Guengerich, F. P. (2006) Proc. Nat. Acad. Sci. USA 103, 13565-13566
Hansmann, U. H. E. (1997) Chem. Phys. Lett. 281, 140-150
Hritz, J., de Ruiter, A., Oostenbrink, C. (2006) J. Med. Chem. (2008) 51, 7469-7477
Karplus, M., Kuriyan, J. (2005) Proc. Nat. Acad. Sci. USA 102, 6679-6685
Keizers, P. H. J., De Graaf, C., de Kanter, F. J. J., Oostenbrink, C., Feenstra, K. A., Commandeur, J. N. M., Vermeulen, N. P. E. (2005) J. Med. Chem. 48, 6117-6127
Stjernschantz, E., van Vugt-Lussenburg, B. M. A., Bonifacio, A., de Beer, S. B. A., van der Zwan, G., Gooijer, C., Commandeur, J. N. M., Vermeulen, N. P. E., Oostenbrink, C. (2008) Proteins 71, 336-352
Stortelder, A., Keizers, P. H. J., Oostenbrink, C., De Graaf, C., De Kruijf, P., Vermeulen, N. P. E., Gooijer, C., Commandeur, J. N. M., van der Zwan, G. (2006) Biochem. J. 393, 635-643
Sugita, Y., Kitao, A., Okamoto, Y. (2000) J. Chem. Phys. 113, 6042-6051
Tasca, F., Gorton, L., Harreither, W., Haltrich, D., Ludwig, R., Nöll, G. (2009) Anal. Chem. 81, 2791-2798
van den Bosch, M., Swart, M., Snijders, J. G., Berendsen, H. J. C., Mark, A. E., Oostenbrink, C., van Gunsteren, W. F., Canters, G. W. (2005) ChemBioChem. 6, 738-746
van Gunsteren, W. F., Bakowies, D., Baron, R., Chandrasekhar, I., Christen, M., Daura, X., Gee, P., Geerke, D., Glättli, A., Hünenberger, P. H., Kastenholz, M. A., Oostenbrink, C., Schenk, M., Trzesniak, D., van der Vegt, N. F. A., Yu, H. B. (2006) Angew. Chem. Intnl. Ed. 45, 4064-4092